Technology
We have created an original technology for development of new chemical compounds, based on numerical methods, organic synthesis, measurement of their biological activity and testing on experimental animals. The main characteristic feature of this technology is careful and extrimally correct control of any experimental activity by numerical methods with instant feedback.
Modern approach to development of new drugs lies in understanding of the disease mechanism, determination of the target protein involved in chemical transformations associated with this disease and inhibiting of this target protein by a chemical-drug. In order for the substance to inhibit a target protein, it must interact with this protein. The degree of interaction is determined by the inhibition constant, the smaller it is, the better. In modern drugs, the inhibition constant is less than 100 nM (nano moles) usually.
Synthesis of new chemicals - potential drugs - is a very expensive process and not every substance inhibits a given target protein. Therefore, it becomes necessary to theoretically or numerically predict the inhibition constant of any substances for a given target protein. A correct theoretical prediction can save a significant amount of time and money during development of new drugs.
Our studies have shown that to date the accuracy of known methods for prediction of inhibition constants in several orders is not very suitable for successful search for a substance with good biological activity. For these reasons, we have developed our own, original methods for prediction of interaction of molecules with proteins. The main idea of these methods is combination of methods based on physical models and methods using experimental data. At the initial stage of search for new drugs, a method based on physical models is used, and, in the course of synthesis and measurement of biological activity of synthesized substances, with further accumulation of experimental data during work, these data are used to correct the physical model in order to improve its predictive ability. What essentially distinguishes our method from all known others is that for training it uses not only positive results (when substances are active), but also negative (when substances are not active) to the same extent. The numerical experiments carried out by us have shown that this approach significantly improves the accuracy of inhibition constant prediction.
Our numerical methods, designed to predict the interaction of molecules with proteins, are implemented in the form of programs and some of these methods are freely available as the ScoreL 1.0 program.
The number of chemicals - potential drugs is 35! = 1040, it is a colossal number! The number of known synthesized substances is only about 108, which is much less than the number of potential drugs. Therefore, to successfully search for biologically active substances, new compounds must be synthesized. However, chemical synthesis is an extremely expensive activity. For this reason, we have developed such an approach to the search for drugs, when possibilities of chemical synthesis are directly taken into account in theoretical and numerical calculations. The most optimal synthesis scheme is selected for given target protein and potential drug with regard to its cost and feasibility, and theoretical calculations are carried out within the framework of this scheme, which significantly saves time and money.
Our experimental laboratory is equipped with the equipment necessary for organic synthesis and purification of substances. Currently we manage the following synthesis methods:
- nucleophilic substitution in aromatic and aliphatic carbon atoms (including the amide bond formation);
- electrophilic substitution in aromatic carbon atom;
- reaction of the double C = C bond formation;
- organoelemental synthesis: organophosphorus (a method of phosphorylation, synthesis of triphosphates), sulfur-containing compounds;
- formation of С-С bonds by means of organometallic reagents (Grignard reagent);
- synthesis of heterocyclic compounds;
- oxidation and reduction reactions (for example, esters into alcohols, nitro group to amino group, azides to amino group), as well as other modifications of functional groups.
In order to measure biological activity of chemical compounds, potential drugs-anticoagulants in vitro, we have purchased necessary equipment and reagents, and also mastered the measurement procedure.
On the basis of the Russian Cardiology Research and Production Complex, we have organized preclinical trials of anticoagulants on laboratory rats of lines Wistar-Kyoto (WKY) and/or Spontaneously hypertensive rats (SHR).
Based on the results of our trials, two applications for a patent have been filed:
- “Method for selecting potential medicinal compounds” (WO/2006/110064) – has been transferred to the national phase in the USA.
- “Method of determination of protein ligand binding and of the most probable ligand pose in protein binding site” (WO/2008/127136)
The following article was published:
Tarasov D.N., Tovbin D.G., "How sophisticated should a scoring function be to ensure successful docking, scoring and virtual screening?" Journal of Molecular Modeling, 2009 (15) 329-341.
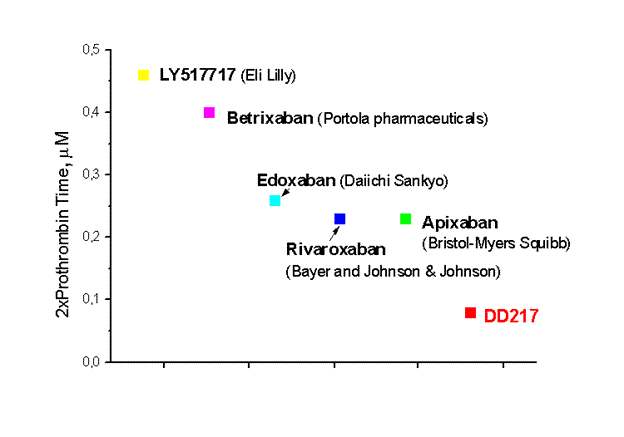
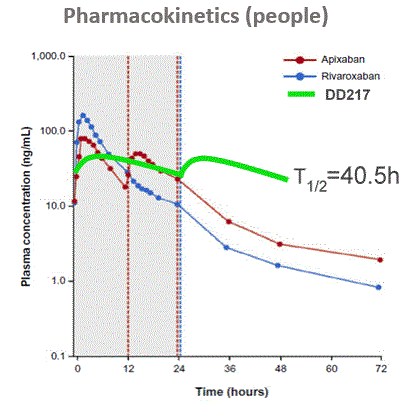
- The DD217 compound half-life in humans is 40.5 hours compared to 8-10 hours for the main competitors - Rivaroxaban and Apixaban. More “soft” long-term action in comparison with competitors leads to a better safety profile and marketing advantages in the form of convenience of consumption. In the result, we have a candidate for the best in class among oral anticoagulants and the blockbuster potential.